
A las matemáticas se les suele llamar el "lenguaje de la ciencia". Es muy adecuado para el procesamiento cuantitativo de casi cualquier información científica, independientemente de su contenido. Y con la ayuda del formalismo matemático, los científicos de diferentes campos pueden, hasta cierto punto, "entenderse" entre sí. Hoy en día, está surgiendo una situación similar con la informática. Pero si las matemáticas son el lenguaje de la ciencia, entonces CS es su navaja suiza. De hecho, es difícil imaginar la investigación moderna sin analizar y procesar grandes cantidades de datos, cálculos complejos, modelado por computadora, visualización y el uso de software y algoritmos especiales. Veamos algunas "historias" interesantes cuando diferentes disciplinas utilizan métodos de informática para resolver sus problemas.
Bioinformática: de las placas de Petri a la biología In silico
La bioinformática puede considerarse uno de los ejemplos más llamativos de la intersección de la informática y otras disciplinas. Esta ciencia se ocupa del análisis de datos biológicos moleculares mediante métodos informáticos. La bioinformática como una dirección científica separada apareció a principios de los 70 del siglo pasado, cuando se publicaron por primera vez secuencias de nucleótidos de ARN pequeños y se crearon algoritmos para predecir su estructura secundaria (la disposición espacial de los átomos en una molécula).
Ha comenzado una nueva era de la bioinformática con el Proyecto Genoma Humano, que tiene como objetivo determinar la secuencia de nucleótidos en el ADN humano e identificar genes en el genoma. El costo de la secuenciación del ADN (secuenciación de nucleótidos) se ha reducido en varios órdenes de magnitud. Esto ha llevado a un tremendo aumento en el número de secuencias en las bases de datos públicas. El siguiente gráfico muestra el crecimiento en el número de secuencias en la base de datos pública de GenBank desde diciembre de 1982 hasta febrero de 2017 en una escala semilogarítmica. Para que los datos acumulados sean útiles, deben analizarse de alguna manera.

Crecimiento en el número de secuencias en GenBank desde diciembre de 1982 hasta febrero de 2017. Fuente: www.ncbi.nlm.nih.gov/genbank/statistics
Uno de los métodos de análisis de secuencias en bioinformática es la alineación de secuencias. La esencia del método radica en el hecho de que las secuencias de monómeros de ADN, ARN o proteínas se colocan una debajo de la otra de manera que se vean áreas similares. La similitud en las estructuras primarias (es decir, secuencias) de dos moléculas puede reflejar su relación funcional, estructural o evolutiva. Dado que una secuencia se puede representar como una cadena con un alfabeto específico (4 nucleótidos para el ADN y 20 aminoácidos para la proteína), la alineación resulta ser una tarea combinatoria de CS (por ejemplo, la alineación de líneas también se usa en el procesamiento del lenguaje natural - PNL). Sin embargo, el contexto de la biología agrega cierta especificidad al problema.
Veamos la alineación usando proteínas como ejemplo. Un residuo de aminoácido en la proteína corresponde a una letra del alfabeto latino en la secuencia. Las cadenas están escritas una debajo de la otra para lograr la mejor coincidencia. Los elementos coincidentes están uno debajo del otro, los "espacios" se reemplazan por "-" (espacio). Designan indel , es decir, el lugar de la posible inserción (introducción en una molécula de uno o más nucleótidos o aminoácidos) y deleciones ( "deserción" de un nucleótido o amino ácido).

Un ejemplo del alineamiento de las secuencias de aminoácidos de dos proteínas. La leucina (L) y la isoleucina (I), que son isómeros, están resaltadas en azul; esta sustitución en la mayoría de los casos no afecta la estructura de la proteína.
Sin embargo, ¿cómo se puede determinar si la alineación es óptima? Lo primero que me viene a la mente es estimar el número de coincidencias: cuantas más coincidencias, mejor. Sin embargo, en el contexto de la biología, esto no es del todo cierto. Las sustituciones (sustituciones de un aminoácido por otro) son desiguales: algunas sustituciones (por ejemplo, S y T, D y E son residuos que difieren en estructura exactamente en un átomo de carbono) prácticamente no afectan la estructura de las proteínas. Pero reemplazar la serina con triptófano cambiará en gran medida la estructura de la molécula. Se ingresa un criterio cuantitativo (ponderación o puntuación) para determinar si la compensación es la mejor posible. Para evaluar las sustituciones se utilizan las denominadas matrices de sustitución, basadas en las estadísticas de sustitución de aminoácidos en proteínas de estructura conocida. Cuanto mayor sea el número en la intersección de las letras coincidentes, mayor será la puntuación.

Periódicamente aparecen nuevas matrices de sustitución. Aquí está la matriz BLOSUM62.La
puntuación también tiene en cuenta la presencia de deleciones. Por lo general, la penalización por "abrir" una eliminación es varios órdenes de magnitud mayor que por "continuar". Esto se debe al hecho de que una sección de varios huecos consecutivos se considera una mutación, y varios huecos en diferentes lugares se consideran varios. En el siguiente ejemplo, el primer par de secuencias es más similar que el segundo, porque en el primer caso, las secuencias están formalmente separadas por un evento evolutivo:

Ahora sobre los propios algoritmos de alineación. Hay dos tipos de alineación emparejada (encontrar áreas similares de dos secuencias): global y local. El alineamiento global implica que las secuencias son homólogas (similares) en toda su longitud. Incluye ambas secuencias en su totalidad. Sin embargo, con este enfoque, áreas similares no siempre están bien definidas si son pocas. La alineación local se utiliza si las secuencias se mantienen como homólogas (por ejemplo, debido a la recombinación) y sitios no relacionados. Pero no siempre puede entrar en el área de interés, además, existe la posibilidad de encontrar un área similar accidental. Para obtener una alineación por pares, se utilizan métodos de programación dinámica (resolver un problema dividiéndolo en varias subtareas idénticas conectadas de forma recurrente). En los programas para la alineación global, el algoritmo de Needleman-Wunsch se utiliza a menudo , y para la alineación local, el algoritmo de Smith-Waterman . Puede leer más sobre ellos siguiendo los enlaces.
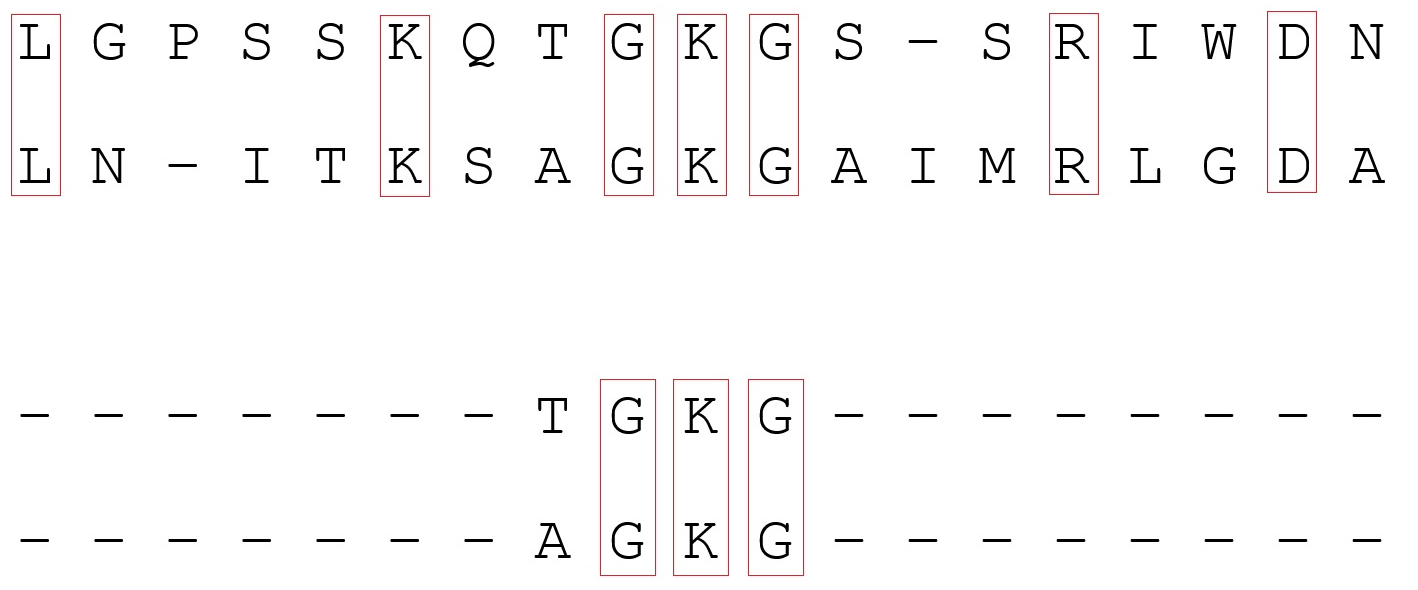
Ejemplo de alineación: la parte superior es global, la inferior es local. En el primer caso, la alineación se produce a lo largo de toda la longitud de las secuencias; en el segundo, se encuentran algunas regiones homólogas.
Como puede ver, la tarea biológica puede reducirse bastante a la tarea de CS. La alineación por pares utilizando los algoritmos mencionados requiere aproximadamente m * n de memoria adicional (m, n son las longitudes de las secuencias), que las computadoras hogareñas modernas pueden manejar fácilmente. Sin embargo, la bioinformática también tiene tareas más no triviales, por ejemplo, alineación múltiple (alineación de varias secuencias) para la reconstrucción de árboles filogenéticos.... Incluso si comparamos 10 proteínas muy pequeñas con una longitud de secuencia de aproximadamente 100 caracteres, se requerirá una cantidad inaceptablemente grande de memoria adicional (la dimensión de la matriz es 100 ^ 10). Por lo tanto, en este caso, la alineación se basa en varias heurísticas.
Modelando la estructura a gran escala del universo
A diferencia de la biología, la física ha ido de la mano de la informática desde los primeros días de las computadoras. Antes de la creación de las primeras computadoras, la palabra "computadora" (calculadora) se llamaba una posición especial: eran personas que realizaban cálculos matemáticos en calculadoras. Así, durante el Proyecto Manhattan, el físico Richard Feynman fue el director de todo un equipo de "calculadoras" que procesaban ecuaciones diferenciales en máquinas sumadoras.

"Sala de Computación" del Centro de Investigación de Vuelo. Armstrong. EE.UU., 1949
En la actualidad, los métodos de CS se utilizan ampliamente en varios campos de la física. Por ejemplo, la física computacional estudia algoritmos numéricos para resolver problemas físicos para los que ya se ha desarrollado una teoría cuantitativa. En situaciones en las que la observación directa de objetos es difícil (esto sucede a menudo en astronomía), el modelado informático ayuda a los científicos. Exactamente tal caso es el estudio de la estructura a gran escala del Universo : las observaciones de objetos distantes son difíciles debido a la absorción de radiación electromagnética en el plano de la Vía Láctea, por lo que el modelado se ha convertido en el principal método de investigación.

,
Una de las tareas de la cosmología moderna es explicar la imagen observada de la diversidad de galaxias y su evolución. A nivel cualitativo, ya se conocen los procesos físicos que ocurren en las galaxias, por lo que los esfuerzos de los científicos están encaminados a obtener predicciones cuantitativas. Esto permitirá responder a una serie de preguntas fundamentales, por ejemplo, sobre las propiedades de la materia oscura. Pero, antes de aislar las manifestaciones observadas de la materia oscura, es necesario comprender el comportamiento de la materia ordinaria. A gran escala (varios millones de años luz), la materia ordinaria se comporta efectivamente de la misma manera que la oscuridad: está sujeta a una fuerza de gravedad, puede olvidarse de la presión del gas. Esto hace que sea relativamente fácil simular la evolución de la estructura a gran escala del Universo (métodos numéricos,que contiene solo materia oscura o similar al polvo y que reproduce bien la estructura a gran escala de la distribución de las galaxias, comenzó a desarrollarse desde la década de 1980).
La materia oscura se modela de la siguiente manera. El cubo virtual, de cientos de millones de años luz de tamaño, está casi uniformemente lleno de partículas de prueba: cuerpos. Desde el principio, en el Universo se presentaron pequeñas inhomogeneidades, de las cuales surgió toda la estructura observada, por lo que el relleno es "casi uniforme". Entonces las partículas comienzan a “vivir sus propias vidas” bajo la influencia de la gravedad: el problema de los N cuerpos está resuelto . Las partículas que se escapan del cubo se transfieren a la cara opuesta y las fuerzas gravitacionales también se propagan con la transferencia. Gracias a esto, el cubo se vuelve, por así decirlo, infinito, como el universo.

Trayectorias aproximadas de tres cuerpos idénticos ubicados en los vértices de un triángulo no isósceles y que tienen velocidades iniciales nulas
Uno de los modelos numéricos más famosos de este tipo es el Millenium , que tiene un tamaño de cubo de más de 1.500 millones de años luz y unos 10 mil millones de partículas. En los años siguientes, se fabricaron varios modelos más grandes: el Horizon Run con un lado del cubo 4 veces más grande que el Millenium, y el Dark Sky con 16 veces el Millenium. Estos y modelos similares han jugado un papel clave en proyectos para validar el modelo Lambda-CDM ahora generalmente aceptado. (Un universo que contiene aproximadamente un 70% de energía oscura, un 25% de materia oscura y un 5% de materia ordinaria).

Millenium: , ; — . .
La reducción de escala causa problemas al hacer coincidir observaciones y modelos numéricos con una materia oscura. En una escala más pequeña (la escala de propagación de las ondas de choque de las supernovas), la materia ya no puede considerarse polvorienta. Es necesario tener en cuenta la hidrodinámica, el enfriamiento y calentamiento del gas por radiación, y mucho más. Para tener en cuenta todas las leyes de la física en el modelado, se realizan algunas simplificaciones: por ejemplo, puede dividir el cubo modelo en una red de celdas (física de subred) y asumir que cuando se alcanza una cierta densidad y temperatura en la celda , parte del gas se convertirá instantáneamente en una estrella. Esta clase de modelos incluye los proyectos EAGLE e illustris . Uno de los resultados de estos proyectos es la reproducción de la relación Tully-Fisher entre la luminosidad de la galaxia y la velocidad de rotación del disco.
Lingüística y aprendizaje automático: un paso más cerca de resolver un misterio de 4.000 años
Los métodos de CS encuentran aplicaciones en áreas más inesperadas, por ejemplo, en el estudio de lenguajes y sistemas de escritura antiguos. Así, un estudio de un grupo de científicos dirigido por Rajesh P.N. Rao, profesor de la Universidad de Washington, arrojó luz sobre el misterio de la escritura del valle del Indo.
La escritura del Indo, utilizada entre el 2600 y el 1900 a. C. en lo que hoy es Pakistán Oriental y el noroeste de la India, pertenecía a una civilización no menos compleja y misteriosa que sus contemporáneas mesopotámicas y egipcias. Quedan muy pocas fuentes escritas de él: los arqueólogos han encontrado solo alrededor de 1.500 inscripciones únicas en fragmentos de cerámica, tabletas y sellos. La letra más larga tiene solo 27 caracteres.

Inscripciones en focas del valle del Indo
En la comunidad científica, existían varias hipótesis sobre los "símbolos misteriosos". Algunos expertos consideraban que los símbolos no eran más que simples "imágenes bonitas". Entonces, en 2004, el lingüista Steve Farmer publicó un artículo en el que se argumentó que la escritura del Indo no es más que símbolos políticos y religiosos. Su versión, aunque controvertida, todavía encontró partidarios.
Rajesha P.N. Rao, un científico de aprendizaje automático, leyó sobre la escritura hindú en la escuela secundaria. Un grupo de científicos bajo su liderazgo decidió realizar un análisis estadístico de los documentos confiables existentes. En el curso de la investigación utilizando cadenas de Markov(una de las primeras disciplinas en las que las cadenas de Markov encontraron aplicación práctica fue la crítica textual) se comparó la entropía condicionalsímbolos de la escritura del Indo con la entropía de secuencias de signos lingüísticos y no lingüísticos. La entropía condicional es la entropía de un alfabeto para el que se conocen las probabilidades de una letra tras otra. Se seleccionaron varios sistemas para compararlos. Los sistemas lingüísticos incluían: escritura logográfica sumeria, antiguo tamil abugida, sánscrito del Rig Veda, inglés moderno (las palabras y letras se estudiaron por separado) y el lenguaje de programación Fortran. Los sistemas no lingüísticos se dividieron en dos grupos. El primero incluía sistemas con un orden rígido de signos (conjunto artificial de signos n. ° 1), el segundo - sistemas con un orden flexible (proteínas de bacterias, ADN humano, conjunto artificial de signos n. ° 2). Como resultado, resultó que la escritura protoindia resultó ser moderadamente ordenada, como la escritura de las lenguas habladas:la entropía de los documentos existentes es similar a la entropía de las escrituras sumeria y tamil.

Entropía condicional para varios sistemas lingüísticos y no lingüísticos
Este resultado refutó la hipótesis sobre el uso ornamental de los signos. Y aunque los métodos de CS ayudaron a confirmar la versión de que los símbolos del valle del Indo son probablemente un sistema de escritura, el asunto aún no se ha descifrado.
Conclusión
Por supuesto, muchas áreas en las que los métodos de CS encuentran aplicación están por la borda. Es simplemente imposible en un artículo revelar cómo la ciencia moderna se basa en la tecnología informática. Sin embargo, espero que los ejemplos dados muestren cómo se pueden resolver diferentes problemas, incluso mediante métodos CS.
Los servidores en la nube de Macleod son rápidos y seguros.
Regístrese usando el enlace de arriba o haciendo clic en el banner y obtenga un 10% de descuento durante el primer mes de alquiler de un servidor de cualquier configuración.
