
Una proteína de la bacteria Staphylococcus aureus
A finales de noviembre, el equipo DeepMind de Google anunció que su sistema de aprendizaje profundo AlphaFold había alcanzado niveles de precisión sin precedentes en la resolución del problema del plegamiento de proteínas , un problema difícil de la bioquímica computacional.
¿Cuál es el problema y por qué es tan difícil de resolver?
Las proteínas son largas cadenas de aminoácidos. Su ADN codifica estas secuencias y el ARN ayuda a producir proteínas de acuerdo con este modelo genético. Las proteínas se sintetizan en forma de cadenas lineales, pero posteriormente se pliegan en estructuras esféricas complejas (ver la imagen al principio del artículo).
Una parte de la cadena se puede enrollar en una espiral cerrada " α-helix . "La otra parte puede doblarse hacia adelante y hacia atrás para formar una figura ancha y plana," β-sheet ":
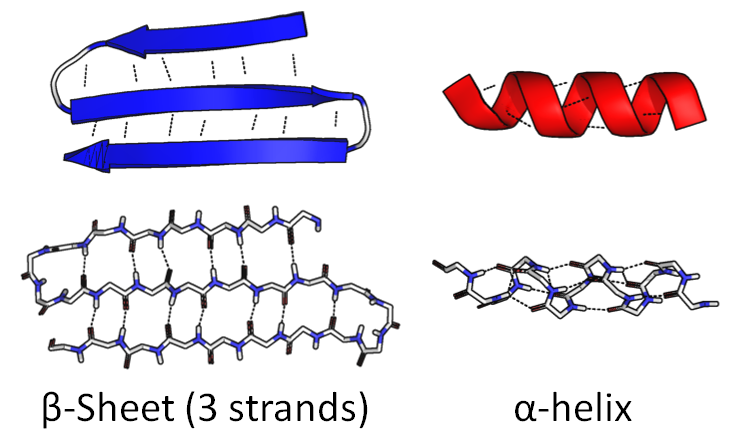
La secuencia de aminoácidos en sí misma se llama estructura primaria . Estas figuras se denominan estructura secundaria .
Estos componentes también se pliegan para formar formas complejas únicas. Esto se llama estructura terciaria :
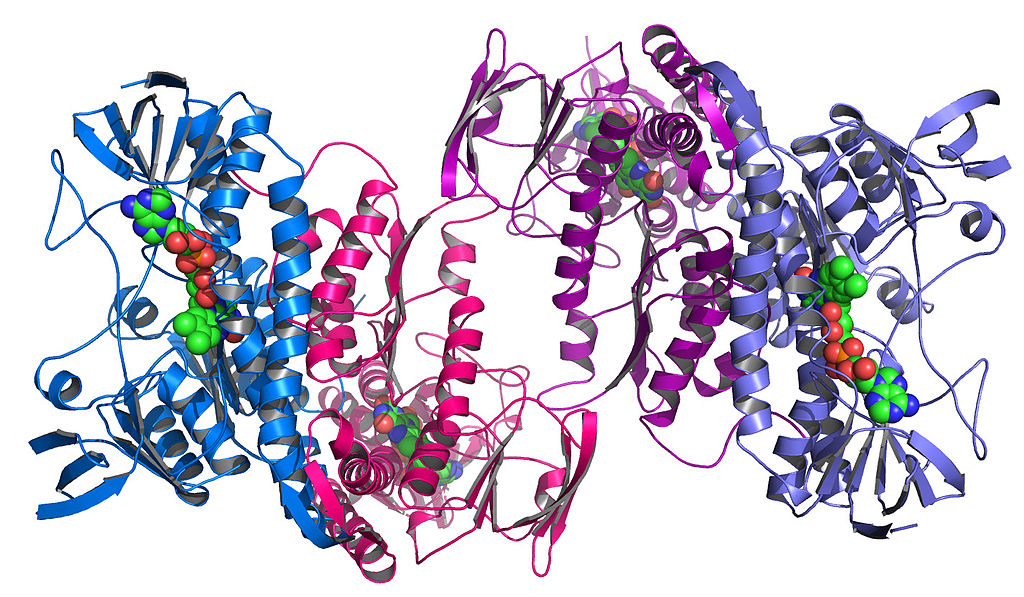
una enzima extraída de la bacteria Colwellia psychrerythraea

proteína RRM3
Parece desordenada. ¿Por qué es tan importante esta bola enredada de aminoácidos?
¡La estructura de las proteínas no es aleatoria! Cada proteína se pliega en una estructura distinta, única y en gran parte predecible, que es esencial para que funcione correctamente. Debido a su forma física, la proteína se adapta bien a las estructuras con las que puede unirse. También son importantes otras propiedades físicas, especialmente la distribución de la carga eléctrica sobre la proteína. En la imagen, la carga positiva se indica en azul, la carga negativa en rojo:

Distribución de la carga superficial en la proteína transportadora de lípidos de las plantas 1 de arroz
Si una proteína es, en esencia, una nanomáquina autoensamblada, entonces el propósito principal de una secuencia de aminoácidos será producir su forma única, distribución de carga y todo lo demás que determina la función de la proteína. Cómo se lleva a cabo exactamente este proceso aún no está del todo claro; hoy en día es un área activa de investigación.
En cualquier caso, comprender la estructura es importante para comprender cómo funciona. Sin embargo, la secuencia de ADN solo define la estructura primaria de la proteína. ¿Cómo conocemos sus estructuras secundarias y terciarias, es decir, la forma exacta que tomará esta maraña?
Este problema se denomina problema de plegamiento de proteínas y existen dos enfoques básicos: medición y predicción.
Los métodos experimentales pueden medir la estructura de una proteína. Sin embargo, esto no es tan fácil de hacer: las estructuras no son visibles a través de un microscopio óptico. Durante mucho tiempo, la cristalografía de rayos X fue el método principal para estudiar estructuras. Además, se utilizó la resonancia magnética nuclear y recientemente ha aparecido una nueva tecnología, la microscopía crioelectrónica .

Patrón de difracción de rayos X de la proteasa del SARS
Sin embargo, estos métodos son costosos, complejos y requieren mucho tiempo y, además, no funcionan con todas las proteínas. En particular, las proteínas incrustadas en la membrana celular, el mismo receptor de la enzima convertidora de angiotensina 2 (ACE2) al que se une el virus COVID-19, se pliega en la bicapa lipídica.células, y es muy difícil de cristalizar.

La estructura de la membrana celular
Por lo tanto, pudimos desmontar la estructura de un pequeño porcentaje de las proteínas secuenciadas . La base de datos de proteínas universal contiene 180 millones de secuencias, mientras que la base de datos de estructuras de proteínas tridimensionales contiene solo 170 mil posiciones.
Necesitamos un método mejor.
* * *
Recordemos que las estructuras secundarias y terciarias de las proteínas son básicamente una función de la estructura primaria que conocemos a través de la secuenciación. ¿Y si, en lugar de medir la estructura de una proteína, pudiéramos predecirla?
Esta es la tarea de predecir la estructura de las proteínas. Los bioquímicos computacionales han estado trabajando en él durante décadas.
¿Cómo puedes abordarlo?
La forma obvia es simular la física del proceso directamente. Simulamos fuerzas para cada átomo, teniendo en cuenta su ubicación, carga y enlaces químicos. Contamos las aceleraciones y velocidades y, paso a paso, nos desplazamos por la evolución del sistema. A esto se le llama "dinámica molecular".

Supercomputadora " Anton " de DE Shaw Research

Supercomputadora IBM Blue Gene
Online puzzle Foldit
El problema es que este enfoque es extremadamente intensivo en computación. Una proteína típica contiene cientos de aminoácidos, es decir, miles de átomos. El medio ambiente también es importante: al plegarse, la proteína interactúa con el agua circundante. Por tanto, es necesario simular el comportamiento de unos 30 mil átomos. En este caso, se produce una interacción electrostática entre cada par de átomos, es decir, con una estimación aproximada, obtenemos 450 millones de pares, un problema de complejidad O (N2). Existen algoritmos inteligentes que reducen su complejidad a O (N log N). Además, para la simulación, es necesario calcular 10 9 -10 12 pasos. Dolor de cabeza excepcional.
De acuerdo, pero no necesitamos simular todo el proceso de plegado. Otro enfoque sugiere encontrar una estructura con energía potencial mínima. Por lo general, los objetos tienden a detenerse con la menor cantidad de energía, por lo que este enfoque heurístico está justificado. La energía se puede calcular mediante el mismo modelo de dinámica molecular, lo que nos da la magnitud de las interacciones. Con este enfoque, podemos probar un grupo de candidatos y elegir la estructura con la menor energía. El problema, por supuesto, es de dónde sacar las estructuras. Simplemente hay demasiados: el biólogo molecular Cyrus Levintol ha calculado que puede haber alrededor de 10,300 . Naturalmente, puede utilizar un enfoque más inteligente que la fuerza bruta aleatoria. Pero todavía hay demasiados.
Por lo tanto, ya se han hecho muchos intentos para acelerar dichos cálculos. Anton, una supercomputadora de DE Shaw Research, utiliza equipos especiales: circuitos integrados especiales. IBM también está utilizando la supercomputadora biológica Blue Gene. Stanford lanzó el proyecto Folding @ Home, utilizando el poder distribuido de las computadoras domésticas. El proyecto Foldit de UW convirtió el plegado en un juego para agregar intuición humana a la computación.
Sin embargo, durante mucho tiempo, ninguna tecnología ha sido capaz de predecir una amplia gama de estructuras de proteínas con gran precisión. En la competencia CASP que se realiza dos veces al año, donde los resultados de los algoritmos se comparan con las estructuras medidas experimentalmente, los primeros lugares recibieron predicciones con una precisión del 30-40%. Hasta hace poco:

La mejor precisión predictiva mediana del equipo en la categoría de modelado gratuito
¿Cómo funciona AlphaFold? Utiliza múltiples redes neuronales profundas para aprender diferentes funciones asociadas con cada proteína. Una de las funciones clave es predecir las distancias resultantes entre pares de aminoácidos. Esto lleva el algoritmo a la estructura final. En una variante del algoritmo (descrito en las revistas Nature and Proteins ), se derivó la función potencial de esta predicción, a la que se aplicó el descenso de gradiente más simple, que funcionó sorprendentemente bien.
La principal ventaja de AlphaFold sobre los métodos anteriores es que no necesita hacer suposiciones sobre estructuras. Algunos métodos funcionan dividiendo las proteínas en secciones, contando cada una y luego volviendo a juntar todo. AlphaFold no necesita esto.
Al parecer, DeepMind considera resuelto el problema del plegado, que me parece una simplificación excesiva, pero en cualquier caso, su avance es significativo. Los expertos que no están afiliados a Google usan epítetos como “ fantástico ” y “ revolucionario ”.
La ingeniería genética ahora tiene dos herramientas poderosas, CRISPR y plegamiento de proteínas. Quizás la década de 2020 sea para la biotecnología lo que fue la década de 1970 para la informática.
¡Felicitaciones a los investigadores de DeepMind por este avance!